1. Isolation of Full Length cDNAs
a. Rationale. The goal of the NSF supported 2010 project is to identify the functions of all of the genes in the reference plant Arabidopsis. It is estimated that there are 26,000 in the genome. Identification of all the genes will lead to the identification of the proteins encoded by the genes, and this is of paramount importance for the elucidation of the various signaling pathways as well as for all the other disciplines of Plant Biology. The establishment of a protein database encoded by the Arabidopsis genomic sequence will revolutionize Plant Biology. Two major approaches are currently used for mapping the transcriptional units in the Arabidopsis genomic sequence. (1) ESTs, which are small fragments of mRNAs transcribed from a specific gene and represent only a small portion of the encoded protein. There are approximately 8,000 unique EST sequences from Arabidopsis representing ~40% of the potential transcription units. Annotation of sequences utilizing various programs that predict the presence and the structure of genes across the Arabidopsis genome. Great progress has been made in computational biology during the past 10 years for developing gene prediction tools. The question immediately arises of whether the annotation of the Arabidopsis genome sequence is accurate. The answer is simply `No' because the various programs used by the various sequencing groups cannot accurately predict all the genes and their corresponding structures. The inaccuracy of the annotated sequence is due to the inaccuracy of the software used. For annotation to be precise, it needs to be experimentally verified by the isolation of full-length cDNAs. The task of annotating the entire genome is similar to the historical construction of the map of the North American continent. The first maps were inaccurate because the tools used did not have the precision for fine mapping. As the mapping tools improved, the maps became more accurate. A similar situation exists regarding the annotation of various genomes.
We propose here to construct 15,000 full-length cDNAs identified by global expression studies using chip gene technology. This is the most technically difficult part of our proposal, but its successful completion will allow the isolation of full-length cDNAs for the entire Arabidopsis genome. The full-length cDNAs will be deposited in the Arabidopsis Biological Resource Center to be used by the entire Plant Biology community for protein biochemical studies. We define a full-length cDNA as one that contains part of the UTR (+1) and the polyadenylation site.
b. Isolation of PolyA
d. The UPS cloning system. The availability of the full-length cDNA cloned into these novel vectors will greatly facilitate their use by the Plant Biology community. Recently, a new cloning method was described by Steve Elledge's laboratory (33) that facilitates the rapid and systematic construction of recombinant DNA molecules. The central cloning method is named the univector plasmid-fusion system (UPS). The UPS uses Cre-lox site-specific recombination to catalyze plasmid fusion between the univector - a plasmid containing the gene of interest- and host vectors containing regulatory information. Fusion events are genetically selected and place the gene under the control of new regulatory elements. A second UPS-related method allows for the precise transfer of coding sequences only from the univector into a host vector. The UPS eliminates the need for restriction enzymes, DNA ligases and many in vitro manipulations required for cloning, and allows for the rapid construction of multiple constructs for expression in multiple organisms.
Unlike the conventional 'cut-and-paste' strategy of restriction-enzyme-based methods, recombinant DNA assembled by UPS is achieved by plasmid fusion through site-specific recombination. UPS can be used to fuse a coding region of interest either with a specific promoter to gain novel transcriptional regulation, or with another coding sequence to produce a fusion protein with new properties. UPS eliminates the use of restriction enzymes and DNA ligase: instead, these functions are both carried out simultaneously by a single enzyme, Cre. This relieves the constraints on cloning vectors with respect to DNA sequence and size because the UPS reaction is independent of vector size or sequence. Furthermore, the time-consuming processes inherent in conventional cloning, such as the identification of a suitable vector, designing a cloning strategy, restriction endonuclease digestion, agarose gel electrophoresis, isolation of DNA fragments, and the ligation reaction, is shortened to a 20 minute UPS reaction. Due to the uniformity and simplicity of UPS, dozens of constructs can be made simultaneously by simply using different recipient vectors. In addition, unlike restriction enzymes and DNA ligases, GST-Cre can be made inexpensively in large quantities. These features will save investigators significant amounts of time and expense. Furthermore, these recombination-based technologies open new avenues for the systematic manipulation and processing of large gene sets in parallel, a feature that is essential for future functional genomic research (33).
2. Sequencing the Full Length cDNAs
We propose to isolate and sequence 15,000 full-length cDNAs. The average length of each cDNA is estimated to be 2 kb. Thus, 30 Mb of sequence will be produced during the duration of this grant. Currently, we (SPP consortium) are sequencing the 30 Mb of chromosome 1 of Arabidopsis estimated to be completed sometime between the end of 2000 and the beginning of 2001. The sequencing of full-length cDNAs will be initiated some time at the end of 2000 (a year after the start date of this proposal). Thus, as the SPPC is coming out from the Arabidopsis Genome Sequencing Project, it will get involved with sequencing 30 Mb of full-length cDNAs. This pattern is highly efficient because the human and instrumentation resources that were established for the Arabidopsis genomic sequence will now be utilized for the sequencing of full-length cDNAs.
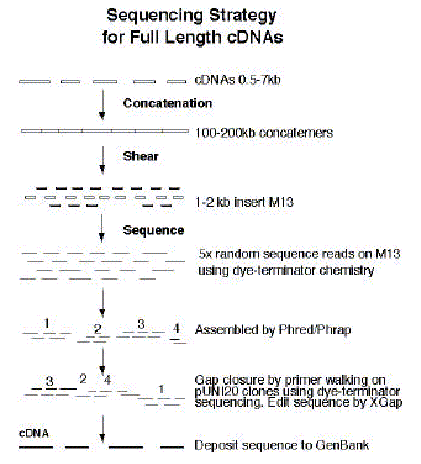
a. Sequencing Strategy Accurate sequence of the 15,000 cDNAs will provide experimental verification of the annotation as well as will allow the precise determination of the molecular mass of each protein encoded by the Arabidopsis genes. To facilitate the rapid, accurate and efficient sequencing of cDNAs we will follow the concatenation cDNA sequencing (CCS) procedure where multiple short DNA molecules 1-2 kb are first ligated to form long DNA fragments or concatemers, that are then randomly sheared and sequenced (56) exactly as currently done for sequencing genomic BAC clones 100 kb in size (PGEC: http://pgec-genome.pw.usda.gov; PENN: http://genome.bio.upenn.edu; Stanford: http://sequence-www.stanford.edu/ara/ArabidopsisSeqStanford.html ). The pUN120 clones will be digested with NotI/SfiI separated by agarose gel electrophoresis and the cDNA inserts will be purified by agarase treatment according to the manufacture conditions. The cDNAs will be ligated to form concatemers. The boundaries between individuals cDNAs are recognized at the stage of computer editing by virtue of the restriction sites that the program used when the clones were first isolated. In our case, they will be NotI/SfiI. The sites are electronically "cut" prior to computer assembly and as a result each cDNA sequence is ultimately identified as an individually assembled contiguous sequence (contig). CCS is an alternative to oligonucleotide walking and deletion library construction (56).
A summary of the sequencing approach is shown below in Fig. 1. This strategy has been very successful for sequencing chromosomes of Arabidopsis.
b. Operation of the SPP Consortium. Each member group of the SPPC performs a unique role to serve the Consortium. Joe Ecker's and Ron Davis's laboratory will send the cDNA clones to A. Theologis' lab, where the shotgun libraries will be constructed in M13. The libraries will be sent to Ron Davis' Genome Center where the libraries are electroporated into E. coli, plaques are picked and grown, and templates are prepared using the high-throughput template preparation robot. Subsequently, the templates are equally distributed among the three laboratories for sequencing. Each site is responsible for a given concatemer, so all the templates prepared from a given clone are sent to the appropriate site, where the sequence is generated, assembled, and submitted to GenBank.
The Consortium will provide the following sequencing capacity towards proposed sequencing: Stanford, U. Penn.
and PGEC.
A MegaBACE capillary sequencer has been requested by PENN and PGEC to replace the 377s after the
Arabidopsis genome-sequencing project is completed.
c. Construction of Sequencing Libraries.
M13 libraries are constructed with concatemer DNA
isolated with a protocol from the MIT Genome Center, which was optimized at the PGEC site. The DNA
is randomly fragmented using the hydrodynamic shearing process developed at the Stanford Center by
Dr. Peter Oefner (38). This method utilizes an HPLC pump with a manifold valve and a stainless steel tee
with recirculation of the DNA sample. The resulting size of the sheared DNA can be changed by varying
the flow rate as the DNA is recirculated through the tee. This shearing procedure has been utilized for the
construction of all 95 M13 libraries used in the Arabidopsis sequencing efforts by the SPPC. The clones
which have been sequenced from these libraries show random distribution of ends and are distributed
relatively evenly across the entire clone/genome. The ends of the sheared DNA are repaired with T4
DNA polymerase and adaptors are ligated containing non-complementary cohesive ends, as diagrammed
in below figure.
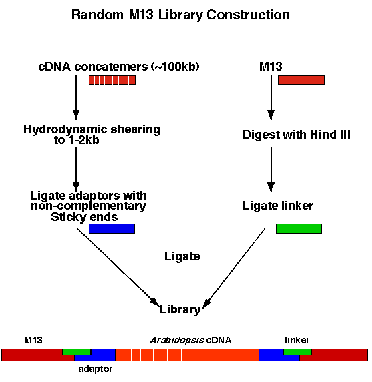
The excess adaptors are removed using agarose gel electrophoresis and the adapted DNA is ligated into a M13 cloning vector. The M13 vector is cleaved with HindIII and a single stranded non-complementary 9-mer (linker) is ligated at the HindIII site generating a complementary end to the cohesive end of the adapted DNA (see Figure 2). This cloning procedure is highly efficient and does not require phosphatasing of the vector. More importantly, it reduces chimera formation that can cause considerable difficulty during sequence assembly.
d. Genome Sequencing Technologies and Strategies. The new instrumentation developed at Stanford for the "front-end" of the sequencing process was designed and constructed with funding from the NCHGR. These instruments have been used at Stanford throughout the current Arabidopsis sequencing project to pick and grow M13 clones and to prepare template DNA for all three sites of the SPP Consortium.
The next step in the sequencing process after DNA template production is preparation of the sequencing reactions. Instrumentation currently in use for this process includes the ABI TurboCatalyst (Stanford & PGEC), which produces one 96-well plate of dye-primer reactions as an ethanol precipitate in ~8 hour, including all pipetting and thermal cycling steps. The alternative method involves setting up the reactions with a multi-channel pipettor manually or with a robot such as the Hamilton, Robbins, or Biomek 2000, transfer to a PE9700 or MJ thermal cycler, followed by purification of the products by ethanol precipitation or column chromatography (all three sites of the SPPC). The latter method is faster than the Catalyst but is more labor-intensive. The sequencing reactions are then run on ABI377 Sequencers.
In the first year of our current sequencing project, most of the shotgun phase of sequencing was carried out using dye primer chemistry with ThermoSequenase (Amersham) and Operon dye-labeled primers. ABI terminator chemistry was used for finishing reactions, since these were 4-5X more expensive than the dye primer reactions. However, recently ABI has released new reagents, using TaqFS and BigDye labeling for both dye primer and dye terminator chemistry. The BigDye reagents are energy-transfer dyes with increased sensitivity, resulting in more reliable detection even with low amounts of template DNA. Amersham has a comparable reagent with their ET-primers, but no comparable terminator chemistry. Also, we have been able to use dilutions of the BigDye chemistry without affecting sequence quality, thereby lowering the cost. Because the cost of the BigDye terminator reactions is now ~25% of the previous cost, we expect to do a higher proportion of the shotgun coverage with dye terminator chemistry. This will also help with throughput requirements since dye terminator reactions are done in a single reaction plate compared to four plates for dye primer chemistry.
Combinations of approaches are being used for finishing the sequences to high accuracy. After assembly of the shotgun data with phredPhrap, if gaps exist, primers are synthesized at the ends of the contigs to walk further on existing templates if possible, or to PCR across the gap, or walk on the plasmid clone. Regions of low quality or low coverage are identified and designated for resequencing with an alternative chemistry. The sequence will be compared to the genomic sequence and if discrepancies exist, the cDNA sequence will be compared with the genomic DNA sequence using denatured HPLC (20,34,39,51,52).
e. Sequence Quantity and Quality. In order to complete 30 Mb of cDNA sequence in 4 years (cDNA sequencing will be initiated during the second year of the project), 7.5 MB of sequence will be produced at the three SPP sites per year or 2.5 Mb/year/site. This corresponds to 75 shotgun libraries/year to be constructed by the PGEC site.
30 Mb sequence produced/4 years = 7.5 Mb/year
5x shotgun coverage x 7.5 Mb = 37.5x10 6 raw bases/year
with an 80% overall success rate, 45x10 6 raw bases/year will be required.
[45x10 6 raw bases/year]/[400 bases/lane] = 112,500 lanes/year
[112,500 lanes/yr]/[48 weeks/yr] = 2344 lanes/week
[2344 lanes/week]/[5 days/wk] = 468 lanes/day
[468 lanes/day]/[96 lanes/gel] = 5 gels/day
[5 gels/day]/3 sites = 2 gels/day/site
The current sequencing capacity of the SPP is 10 gels/day/site.
f. Costs of Production Sequencing. We are requesting a total of $7.5 million (50% of the proposed budget) to complete the sequence of 30 Mb of cDNAs, resulting in a cost of $0.25/base. This cost is based on the actual data obtained during the Arabidopsis sequencing project by the SPPC, which had a cost of $0.50/base, but with 10x coverage. It is lower than that of the genomic sequence because the coverage for the cDNA sequence will be 5x.
3. References:
1. Abel, S. and Theologis, A. 1996. Early genes and
auxin action. Plant Physiol., 111:9-17.
2. Berger, S., Bell, E. and Mullet, J. E. 1996. Two
methyl jasmonate-insensitive mutants show altered
expression of AtVsp in response to methyl jasmonate
and wounding. Plant Physiol., 111:525-531.
3. Bevan, M. and others. 1998. Analysis of 1.9 Mb of
contiguous sequence from chromosome 4 of Arabidopsis
thaliana. Nature, 391:485-488.
4. Bleecker, A. B., Estelle, M. A., Somerville, G. and
Kende, H. 1988. Insensitivity to ethylene conferred
by a dominant mutation in Arabidopsis thaliana.
Science, 241:1086-1089.
5. Boerjan, W., Cervera, M.-T., Delarue, M., Beeckman,
T., Dewitte, W., Bellinin, C., Caboche, M., Van
Onckelen, H., Van Montagu, M. and Inz�, D. 1995.
superroot, a recessive mutation in Arabidopsis,
confers auxin overproduction. Plant Cell,
7:1405-1419.
6. Carninci, P., Kvam, C., Kitamura, A., Ohsumi, T.,
Okazaki, Y., Itoh, M., Kamiya, M., Shibata, K.,
Sasaki, N., Izawa, M., Muramatsu, M., Hayashizaki, Y.
and Schneider, C. 1996. High-efficiency full-length
cDNA cloning by biotinylated cap trapper. Genomics,
37:327-336.
7. Carninci, P., Nishiyama, Y., Westover, A., Itoh, M.,
Nagaoka, S., Sasaki, N., Okazaki, Y., Muramatsu, M.
and Hayashizaki, Y. 1998. Thermostabilization and
thermoactivation of thermolabile enzymes by trehalose
and its application for the synthesis of full length
cDNA. Proc. Natl. Acad. Sci. USA, 95:520-524.
8. Chaudhury, A. M., Letham, S., Craig. S. and Dennis,
E. S. 1993. amp1 - a mutant with high cytokinin
levels and altered embryonic pattern, faster
vegetative growth, constitutive photomorphogenesis
and precocious flowering. Plant J., 4:907-916.
9. Cho, R. J., Campbell, M. J., Winzeler, E. A., Steinmetz, L., Conway, A., Wodicka, L.,
Wolfsberg, T. G., Gabrielian, A. E., Landsman, D., Lockhart, D. J. and Davis, R. W. 1998.
A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell, 2:65-73.
10. Cooke, R., Raynal, M., Laudie, M., Grellet, F.,
Delseny, M., Morris, P-C., Guerrier, D., Giraudat,
J., Quigley, F., Clabault, G., Li, Y-F., Mache, R.
J., Clemnet, B., Philipps, G., Herve, C., Bardet, C.,
Tremousaygue, D., Lescure, B., Lacomme, D., Robey,
D., Jourjon, M-F., Chabrier, P., Charpentwau, J-L.,
Desprez, T., Amselem, J., Chiapello, H. and Hofte, H.
1996. Further progress towards a catalogue of all
Arabidopsis genes: analysis of a set of 5000
no-redundant ESTs. Plant J., 9:101-124.
11. Ecker, J. R. 1995. The ethylene signal
transduction pathway in plants. Science,
268:667-675.
12. Ecker, J. R. 1998. Genes blossom from a weed.
Nature, 391:438-439.
13. Estelle, M. A. and Somerville, C. 1987.
Auxin-resistant mutants of Arabidopsis thaliana with
an altered morphology. Mol. Gen. Genet.,
206:200-206.
14. Fodor, S. P. A., Read, J. L., Pirrung, M. C.,
Stryer, L. and others. 1991. Light-directed,
spatially addressable parallel chemical synthesis.
Science, 251:767-773.
15. Goodman, H., Ecker, J. R. and Dean, C. 1995. The
genome of Arabidopsis thaliana. Proc. Natl Acad.
Sci. USA, 93:10831-10835.
16. Guzman, P. and Ecker, J. R. 1990. Exploiting the
triple response of Arabidopsis to identify
ethylene-related mutants. Plant Cell, 2:513-523.
17. Heller, R. A., Schena, M., Chai, A., Shalon, D. and et al. 1997. Discovery and analysis
of inflammatory disease-related genes using cDNA microarrays. Proc. Natl. Acad. Sci.
USA, 94:8945-8947.
18. H�fte, H., Desprez, T., Amselem, J., Chiapello, H., Caboche, M. and et al. 1993. An
inventory of 1152 expressed sequence tags obtained by partial sequencing of cDNAs from
Arabidopsis thaliana. Plant J., 4:1051-1061.
19. Jacobsen, S. E. and Olszewski, N. E. 1993.
Mutationis at the spindly locus of Arabidopsis alter
gibberellin signal-transduction. Plant Cell,
5:887-896.
20. Jin, L., Underhill, P. A., Oefner, P. J. and
Cavalli-Sforza, L. L. 1995. Systematic search for
polymorphisms in the human genome using denaturing
high-performance liquid chromatography (DHPLC). Am.
J. Hum. Genet., 57:A26.
21. Kakimoto, T. 1996. CKI1, a histidine kinase
homolog implicated in cytokinin signal transduction.
Science, 274:982-985.
22. Karrer, E. E., Lincoln, J. E., Hogenhout, S. and eta l. 1995. In situ isolation of mRNA
from individual plant cells: Creation of cell-specific cDNA libraries. Proc. Natl. Acad.
Sci. USA, 92:3814-3818.
23. Kieber, J. J., Rothenberg, M., Roman, G.,
Feldmann, K. A. and Ecker, J. R. 1993. CTR1, a
negative regulator of the ethylene response pathway
in Arabidopsis, encodes a member of the Raf family of
protein kinases. Cell, 72:427-441.
24. Koornneef, M. and van der Veen, J. H. 1980.
Induction and analysis of gibberellin sensitive
mutants in Arabidopsis thaliana (L.) Heynh. Theor.
Appl. Genet., 58:257-263.
25. Koornneef, M., Elgersma, A., Hanhart, C. J., van
Loenen-Martinet, E. P., van Rijn, L. and Zeevaart, J.
A. D. 1985. A gibberellin insensitive mutant of
Arabidopsis thaliana. Physiol. Plant., 65:33-39.
26. Koornneef, M., Jorna, M. L., Brinkhorst-van der
Swan, D. L. C. and Karssen, C. M. 1982. The isolation
of abscisic acid deficient mutants by selection of
induced revertants in non-germinating gibberellin
sensitive lines of Arabidopsis thaliana (L.) Heynh.
Theor. Appl. Genet., 61:385-393.
27. Koornneef, M., Reuling, G. and Karssen, C. M.
1984. The isolation and characterization of abscisic
acid-insensitive mutants of Arabidopsis thaliana.
Physiol. Plant., 61:377-383.
28. Koshiba, T., Ballas, N., Wong, L.-M. and
Theologis, A. 1995. Transcriptional regulation of
PS-IAA4/5 and PS-IAA6 early gene expression by
indoleacetic acid and protein synthesis inhibitors in
pea (Pisum sativum). J. Mol. Biol., 253:396-413.
29. Lashkari, D. A., DeRisi, J. L., McCusker, J. H.,
Namath, A. F., Gentile, C., Hwang, S. Y., Brown, P.
O. and Davis, R. W. 1997. Yeast microarrays for
genome wide parallel genetic and gene expression
analysis. Proc. Natl. Acad. Sci. USA,
94:13057-13062.
30. Lashkari, D., McCusker, J. and Davis, R. W. 1997. Whole genome analysis:
experimental access to all genome sequenced segments throughlarger-scaled efficient
oligonucleotide synthesis and PCR. Proc. Natl. Acad. Sci. USA, 94:8945-8947.
31. Leyser, O. H. M., Pickett, F. B., Dharmasiri, S.
and Estelle, M. 1996. Mutations in the AXR3 gene of
Arabidopsis result in altered auxin response
including ectopic expression from the SAUR-AC1
promoter. Plant J., 10:403-413.
32. Li, J., Nagpal, P., Vitart, V., McMorris, T. C.
and Chory, J. 1996. A role for brassinosteroids in
light-dependent development of Arabidopsis. Science,
272:398-401.
33. Liu, Q., Li, M. Z., Leibham, D., Cortez, D. and Elledge, S. J. 1998. The univector
plasmid-fusioin system, a method for rapid construction of recombinant DNA without
restriction enzymes. Curr. Biology, 8:1300-1309.
34. Liu, W., Oefner, P., Qian, C., Odom, R. and
Francke, U. 1998. Denaturing HPLC identified novel
FBN1 mutations, polymorphisms and sequence variants.
Am. J. Hum. Genet., in press.
35. Meyerowitz, E. M. 1994. Structure and organization of the Arabidopsis thaliana
nuclear genome. In Arabidopsis, ed. By Meyerowitz, E. M. and Somerville, C. S., Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, Cold Spring Harbor, NY, pp. 21-36.
36. Newman, T., de Bruijin, F. J., Green, P.,
Keegstra, K., Kende, H., McIntosh, L., Ohlrogee, J.,
Raikhel, M., Somerville, S., Thomashow, M., Retzel,
E. and Somerville, C. 1994. Genes galore: a summary
of methods for accessing results fromlarge-scale
partial sequencing of anonymouse Arabidopsis cDNA
clones. Plant Physiol., 106:1241-1255.
37. O'Connell, J. P., Davis, R. W. Chee M. Lockhart D. J. and et al. 1996. After a
period of gestation, DNA chips coming of age. Chem. Eng. News, Dec.:
38. Oefner, P. J., Hunickesmith, S. P., Chiang, L.,
Dietrich, F. and others. 1996. Efficient random
subcloning of DNA sheared in a recirculating
point-sink flow system. Nucleic Acids Res.,
24:3879-3886.
39. Ophoff, R. A., Terwindt, G. M., Vergouwe, M. N.,
van Eijk, R., Oefner, P. J., Hoffman, S. M. G.,
Lamerdin, J. E., Mohrenweiser, H. W., Bulman, D. E.,
Ferrari, M., Haan, J., Lindhout, D., van Ommen, G.-J.
B., Hofker, M. H., Ferrari, M. D. and Frants, R. R.
1996. Familial hemiplegic migraine and episodic
ataxia type-2 are caused by mutations in the Casuper
2+ channel gene CACNL1A4. Cell, 87:543-552.
40. Pickett, F. B., Wilson, A. K. and Estelle, M.
1990. The aux1 mutation of Arabidopsis confers both
auxin and ethylene resistance. Plant Physiol.,
94:1462-1466.
41. Roman, G., Lubarsky, B., Kieber, J. J.,
Rothenburg, M. and Ecker, J. R. 1995. Genetic
analysis of ethylene signal transduction in
Arabidopsis thaliana: Five novel mutant loci
integrated into a stress response pathway. Genetics,
139:1393-1409.
42. Schaefer, B. C. 1995. Revolutions in rapid
amplification of cDNA ends: New strategies for
polymerase chain reaction cloning of full-length cDNA
ends. Analyt. Biochem., 227:255-273.
43. Schena, M., Shalon, D., Davis, R. W and Brown, P.
O. 1995. Quantitative monitoring of gene expression
patterns with a complementary DNA microarray.
Science, 270:467-470.
44. Schena, M., Shalon, D., Heller, R., Chai, A. and et al. 1996. Parallel human genome
analysis: microarray-based expression monitoring of 1,000 genes. Proc. Natl. Acad. Sci.
USA, 93:10614-10619.
45. Seki, M., Carninci, P., Nishiyama, Y., Hayashizaki, Y. and Shinosaki, K. 1998.
High-efficiency cloning of Arabidopsis full-length cDNA by biotinylated CAP trapper.
Plant J., 15:707-720.
46. Shah, J., Tsui, F. and Klessig, D. F. 1997.
Characterization of a salycylic acid-insensitive
mutant (sal1) of Arabidopsis thaliana, identified in
a selection screen itlizing the SA-inducible
expression of the tms2 gene. Mol. Plant Microbe
Inter., 10:69-78.
47. Shoemaker, D. D., Lashkari, D. A., Morris, D. and et al. 1996. Quantitative
phenotypic analysis of yeast deletion mutatnts using a highly parallel molecular
bar-coding strategy. Nature Genetics, 14:Dec.
48. Staswick, P. E., Su, W. and Howell, S. H. 1992.
Methyl jasmonate inhibition of root growth and
induction of a leaf protein are decreased in an
Arabidopsis thaliana mutant. Proc. Natl. Acad. Sci.
USA, 89:6837-6840.
49. Theologis, A., Huynh, T. V. and Davis, R. W. 1985.
Rapid induction of specific mRNAs by auxin in pea
epicotyl tissue. J. Mol. Biol., 183:53-68.
50. Torres-Ruiz, R. A. and Jurgens, G. 1994. Mutations
in the FASS gene uncouple pattern formation and
morphogenesis in Arabidopsis development.
Development, 120:2967-2978.
51. Underhill, P. A., Jin, L., Lin, A. A., Mehdi, S.
Q., Jenkins, T., Vollrath, D., Davis, R. W.,
Cavalli-Sforza, L. L. and Oefner, P. J. 1997.
Detection of numerous Y chromosome biallelic
polymorphisms by denaturing high performance liquid
chromatography (DHPLC). Genome Res., 7:996-1005.
52. Underhill, P. A., Jin, L., Zemans, R., Oefner, P.
J. and Cavalli-Sforza, L. L. 1996. A pre-Columbian
human Y chromosome-specific C -> T transition and its
implications for human evolution. Proc. Natl. Acad.
Sci. USA, 93:196-200.
53. Winzeler, E. A. and Davis, R. W. 1997. Functional analysis of the yeast genome.
Curr. Opinions in Gen. Dev., 7:771-776.
54. Winzeler, E. A., Richards, D. R., Conwat, A. R.,
Goldstein, A. L., Kalman, S., McCullough, M. J.,
McCuskert, J. H., Stevens, D. A., Wodick, L.,
Lockhart, D. J. and Davis, R. W. 1998. Direct allelic
variation scanning of the yeast genome. Science,
281:1191-1197.
55. Wodicka, L., Dong, H., Mittmann, M., Ho, M. H. and
Lockhart, D. J. 1997. Genome-wide expression
monitoring in Saccharomyces cerevisiae. Nature
Biotechnology, 15:1359-1367.
56. Yu, W., Andersson, B., Worley, W. C., Muzny, D. M. and et al. 1997. Large-scale
concatenation cDNA sequencing. Genome Res., 7:353-358.